Quick start (OpenCRAVAT & CHASMplus)¶
The easiest way to obtain CHASMplus scores is by using OpenCRAVAT to fetch precomputed scores.
Install OpenCRAVAT app¶
You can install OpenCRAVAT app for Mac or Windows through the installers provided:
Launch the OpenCRAVAT app, and when prompted install the required system-level annotators to complete the installation process.
Next, click the “Store” tab.
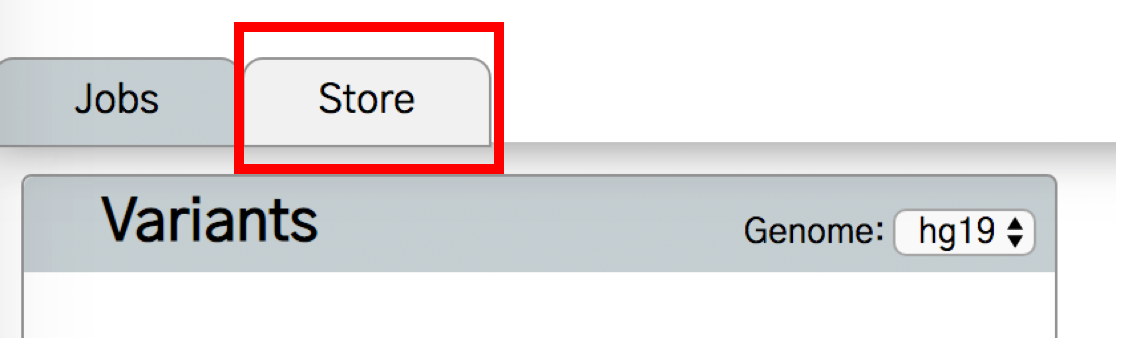
Search for CHASMplus in the store and then click the CHASMplus annotator. You can select either to install the default CHASMplus or a cancer type-specific version.
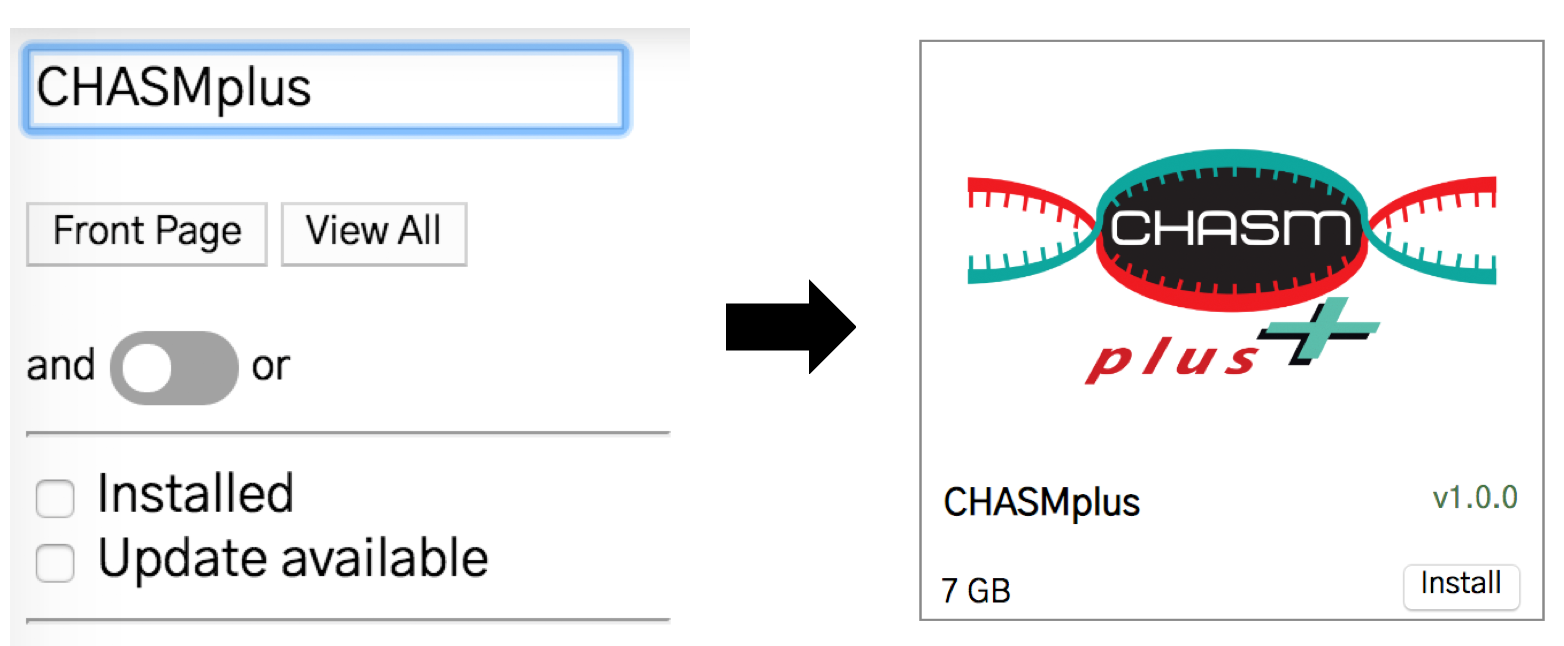
Next, press the “install” button.

It will take several minutes to install, depending on the speed of your internet. By going back to the “jobs” tab in the upper left, you are now ready to submit mutations!
Test example¶
Please download the example input here.
For this example we use a simple tab-delimited format, but OpenCRAVAT can also handle VCF files. The tab-delimited format consists of the following 7 columns: 1) chromosome (with “chr” prefix); 2) Start position (1-based coordinates); 3) Strand (“+” or “-“); 4) Reference allele (“-” for insertions); 5) Alternate allele (“-” for deletions); 6) sample ID; 7) variant ID. For more details about input file formats, please see the OpenCRAVAT wiki.
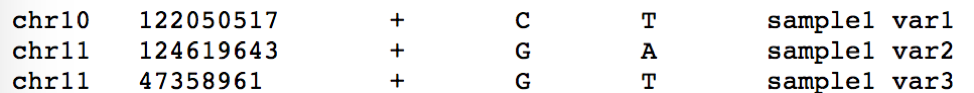
There are four steps to run CHASMplus: 1) upload the input.txt example file (make sure the genome is set to hg38); 2) Make sure CHASMplus is checked in the annotator section; 3) Press the Annotate button in the bottom left; 4) Click to generate an excel output file. CHASMplus results will be on the “variant” tab.
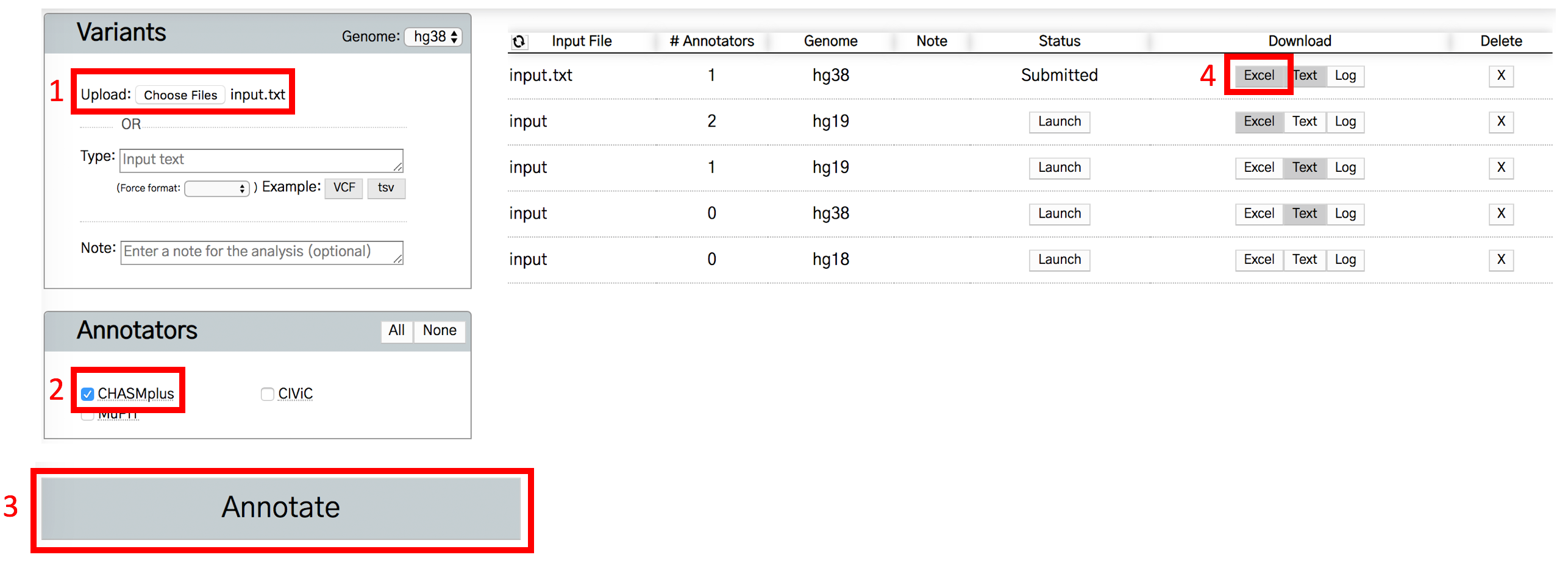
By clicking the “launch” button, you can also interactively explore the results in OpenCRAVAT. Like the excel spreadsheet, CHASMplus results are found on the “variant” tab.
While this tutorial only had you run one annotator (CHASMplus), OpenCRAVAT has 50+ more annotations available.
Interpretation¶
CHASMplus scores range from 0 to 1, with higher scores meaning more likely to be a cancer driver mutation. If you are looking to identify a discrete set of putative driver mutations, then we suggest that you correct the p-values for multiple hypothesis testing. We recommend using the Benjamini-Hochberg (BH) procedure for controling the false discovery rate. You will need to use an external package to do this, e.g., the p.adjust function in R. False discovery rate adjustments will likely be added in the future.
Further documentation¶
For further advanced features of OpenCRAVAT, please see the OpenCRAVAT wiki.
Install Command Line OpenCRAVAT¶
Note
The command line version is meant for users with bioinformatic experience
You will need python 3.6 or newer to use OpenCRAVAT. You will first need to install the OpenCRAVAT python package, please follow the instructions on the OpenCRAVAT wiki:
Install CHASMplus annotators¶
OpenCRAVAT has a modular architecture to perform genomic variant interpretation including variant impact, annotation, and scoring. CHASMplus is one module available in the CRAVAT store. To install the CHASMplus module within OpenCRAVAT, please execute the following command:
$ cravat-admin install chasmplus
The above command may take a couple minutes and will install the pan-cancer model of CHASMplus scores. To install cancer type specific versions of CHASMplus, follow the following template:
$ cravat-admin install chasmplus_LUAD
where LUAD, the abbrevitation from the The Cancer Genome Atlas, designates lung adenocarcinoma. To see a full list of available annotators, issue the following commnad:
$ cravat-admin ls -a -t annotator
Running CHASMplus¶
OpenCRAVAT takes as input either a VCF file or a simple tab-delimited text file. I will describe a simple example that uses the latter. The simple tab-delimited text file should contain a variant ID, chromosome (with “chr”), start position (1-based), strand, reference allele, alternate allele, and optional sample ID.:
chr10 122050517 + C T sample1 var1
chr11 124619643 + G A sample1 var2
chr11 47358961 + G T sample1 var3
chr11 90135669 + C T sample1 var4
chr12 106978077 + A G sample1 var5
You can download an example input file here.
Note
By default, OpenCRAVAT processes variants on the hg38 reference genome. If you are using hg19 or hg18, please specify with the “-l” parameter your specific reference genome so that OpenCRAVAT will know to lift over your variants.
You can run CHASMplus by using the cravat command. For information about command line options, please see the command line help:
$ cravat -h
To obtain CHASMplus scores for pan-cancer (annotator “chasmplus”) and lung adenocarcinoma (annotator “chasmplus_LUAD”), run the following command:
$ cravat -n MYRUN -t excel -a chasmplus chasmplus_LUAD -d output_directory input.txt
The above command will run all annotators (specified by the -a flag, multiple separated by a space) and save results to the directory named “output_directory”. The “-t” option specifies the output to be saved as an excel file. The -n flag specifies the name of the run. Scores and p-values from CHASMplus are found in the “MYRUN.xlsx” file (or “MYRUN.tsv” if -t text is chosen). You should see the “Variant” excel sheet that contains columns like this:
CHASMplus CHASMplus_LUAD
P-value Score Transcript All results P-value Score Transcript All results
0.399 0.048 ENST00000453444.6 ENST00000334433.7:(0.025:0.59),ENST00000358010.5:(0.049:0.393),*ENST00000453444.6:(0.048:0.399),NM_001291876.1:(0.046:0.412),NM_001291877.1:(0.045:0.418),NM_206861.2:(0.048:0.399),NM_206862.3:(0.025:0.59) 0.644 0.013 ENST00000334433.7 *ENST00000334433.7:(0.013:0.644),ENST00000358010.5:(0.023:0.478),ENST00000453444.6:(0.022:0.492),NM_001291876.1:(0.022:0.492),NM_001291877.1:(0.022:0.492),NM_206861.2:(0.023:0.478),NM_206862.3:(0.013:0.644)
0.99 0.001 NM_052959.2 *NM_052959.2:(0.001:0.99) 0.945 0.002 NM_052959.2 *NM_052959.2:(0.002:0.945)
0.446 0.041 NM_001080547.1 ENST00000533968.1:(0.053:0.369),*NM_001080547.1:(0.041:0.446),NM_003120.2:(0.049:0.393) 0.278 0.044 NM_001080547.1 ENST00000533968.1:(0.043:0.284),*NM_001080547.1:(0.044:0.278),NM_003120.2:(0.053:0.224)
CHASMplus scores are provided in a transcript specific manner, with the score for the default selected transcript shown in the “Score”, “P-value”, and “Transcript” columns. Scores for other transcripts are listed in the “All results” column.
Interpretation¶
CHASMplus scores range from 0 to 1, with higher scores meaning more likely to be a cancer driver mutation. If you are looking to identify a discrete set of putative driver mutations, then we suggest that you correct for multiple hypothesis testing. We recommend using the Benjamini-Hochberg (BH) procedure for controling the false discovery rate. You will need to use an external package to do this, e.g., the p.adjust function in R. False discovery rate adjustments will likely be added in the future.
Further documentation¶
For further advanced features of OpenCRAVAT, please see the OpenCRAVAT wiki.